What is phylogeny?
The number of mutations between two organisms can tell us how closely related the two species are. The study of these mutations can also give us an indication of the time that has elapsed since the species' diverged. Comparative biology and the study of homologous traits among species, is relevant in helping to distinguish the different level of relatedness among all organisms [1]. From all of this information, phylogenies and phylogenetic trees may be created to help visualize the evolution of these genetically related group of organisms.
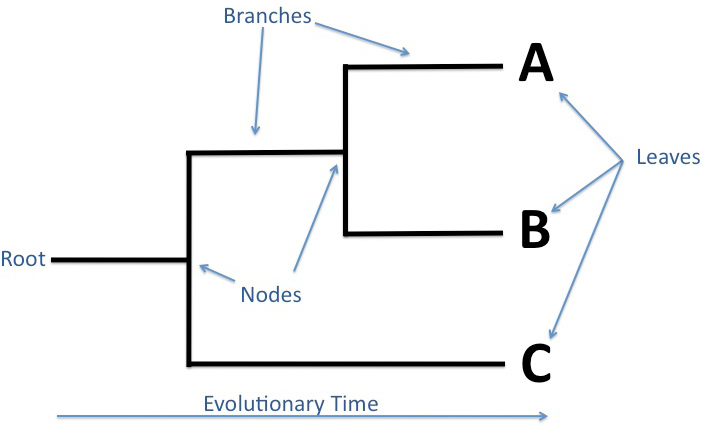
Figure 1. The parts of a phylogenetic tree. The root indicates the common ancestor, while each node represet a more recent common ancestor of the species that branch out from it.
|
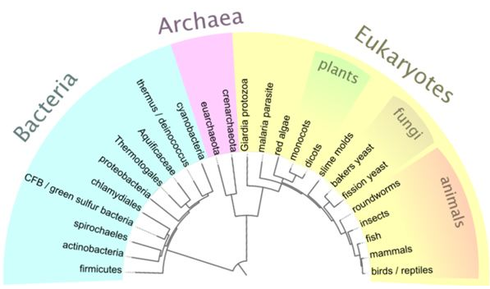
Figure 2. A general cladogram showing the relatedness of many species.
|
What is prkag2's gene phylogeny?
Phylogenetic trees were created using the PRKAG2 gene homologs [as seen on the Gene Homology Page]. These phylogenetic trees were created using ClustalW2 percent identity and BLOSUM62 matrices that help determine the path of evolution with the fewest evolutionary events. Each program uses their own specific algorithm to account for possible insertions and deletions that may happen throughout evolution.
There are two different kind of phylogenetic trees that can be created. An average distance tree uses specific gene sequences in order to create a tree that has the least amount of dissimilarity between its' cluster members, while the neighbor joining tree method produces an unrooted tree that does not require the assumption of a constant rate of evolution [2].
Phylogenetic trees can also be constructed using either a BLOSUM62 or % Identity matrix. The BLOSUM62 matrix uses substitution scores based on the chance that one amino acid would be exchanged for another, while the % Identity matrix uses the actual DNA sequences to determine the amount of similarity between the homologs.
Below are examples of each of the different type and method of phylogenetic tree produced using ClustalW2. The left tree used the average distance tree method, and the right the neighbor joining method, helping to visual the construction differences.
There are two different kind of phylogenetic trees that can be created. An average distance tree uses specific gene sequences in order to create a tree that has the least amount of dissimilarity between its' cluster members, while the neighbor joining tree method produces an unrooted tree that does not require the assumption of a constant rate of evolution [2].
Phylogenetic trees can also be constructed using either a BLOSUM62 or % Identity matrix. The BLOSUM62 matrix uses substitution scores based on the chance that one amino acid would be exchanged for another, while the % Identity matrix uses the actual DNA sequences to determine the amount of similarity between the homologs.
Below are examples of each of the different type and method of phylogenetic tree produced using ClustalW2. The left tree used the average distance tree method, and the right the neighbor joining method, helping to visual the construction differences.
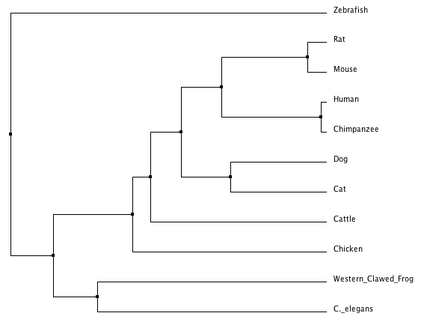
Figure 3. Average distance using % identity (click to enlarge).
|
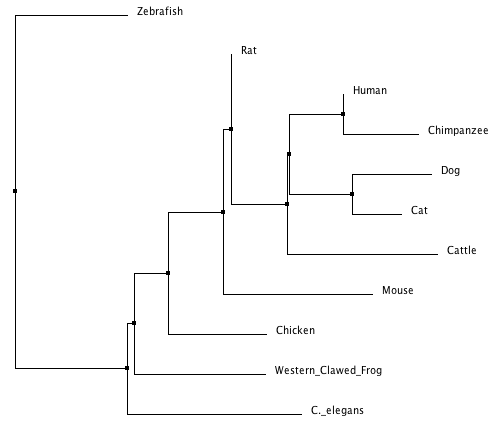
Figure 4. Neighbor joining using % identity (click to enlarge).
|
Analysis
Even though both trees were produced using ClustalW2 and the same matrix (% Identity), they produced a small number of contrasting results. The trees parallel one another in their classification of many of the organisms except for the relationship of how closely related the dog, cat, mouse, and rat gene are to the human homolog. Neighbor joining suggests that the dog and cat are more closely related to the human gene versus the rat and mouse in the average distance tree. For the most part, both trees read quite intuitively that the mammals are most closely related, and that the aquatic species have a different more recent common ancestor. Ultimately, both trees characterize the human prkag2 gene as being closest in relationship to it's chimpanzee homolog.
References
[1] n.d. Plant and Animal Evolution. Retrieved from the University of Waikato School of Science and Engineering Website: http://sci.waikato.ac.nz/evolution/Homology.shtml
[2] Opperdoes, Fred. "The Neighbor-Joining Method." de Duve Institute of Cellular Pathology, ICP. 08 Aug. 1997. Web. 10 May 2013. <http://www.icp.ucl.ac.be/~opperd/private/neighbor.html>
[3] A new bioinformatics analysis tools framework at EMBL-EBI (2010) Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez RNucleic acids research 2010 Jul, 38 Suppl: W695-9 doi:10.1093/nar/gkq313
[2] Opperdoes, Fred. "The Neighbor-Joining Method." de Duve Institute of Cellular Pathology, ICP. 08 Aug. 1997. Web. 10 May 2013. <http://www.icp.ucl.ac.be/~opperd/private/neighbor.html>
[3] A new bioinformatics analysis tools framework at EMBL-EBI (2010) Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez RNucleic acids research 2010 Jul, 38 Suppl: W695-9 doi:10.1093/nar/gkq313
Margaret Beatka ([email protected])
Page Last Updated: 5/10/13
This web page was produced as an assignment for Genetics 677, as an undergraduate course at UW-Madison.
Page Last Updated: 5/10/13
This web page was produced as an assignment for Genetics 677, as an undergraduate course at UW-Madison.
